| Autor |
Nachricht |
Sebi88
Anmeldungsdatum: 05.08.2014
Beiträge: 1
|
 Sebi88 Verfasst am: 05. Aug 2014 13:40 Titel: Quantenmechanische Erklärung der Molekülorbitale Sebi88 Verfasst am: 05. Aug 2014 13:40 Titel: Quantenmechanische Erklärung der Molekülorbitale |
 |
|
Meine Frage:
Hallo zusammen,
ich studiere Chemie mit Nebenfach Physik und habe im Moment leider große Verständisprobleme bei der Entstehung von Molekülorbitalen aus "einfachen" Orbitalen.
Mich beschleicht das Gefühl, ich habe irgendwo einen Verständnisfehler und möchte euch einmal bitten mir zu helfen.
Meine Ideen:
Was mir nicht klar ist, wäre die Frage warum sich z.B. das s-Orbital in ein sigma-s und sigma-s* Orbital aufspaltet.
Wie unterscheidet sich sigma-s von sigma-s* aus quantenmechanischer Sicht?
Für eine Änderung der Orbitale muss meiner Meinung nach auch eine Änderung der Quantenzahlen erfolgen um das neue Orbital zu beschreiben oder nicht?
Vielen Dank schonmal für die Hilfe.
Viele Grüße
Sebi |
|
 |
TomS
Moderator

Anmeldungsdatum: 20.03.2009
Beiträge: 21469
|
| TomS Verfasst am: 05. Aug 2014 14:49 Titel: |
|
|
Nun, letztlich verhält es sich wie folgt: Zunächst löst man die Schrödingergleichung
|\phi\rangle = 0 )
für ein isoliertes Atom, wobei man die inneren Schalen näherungsweise dadurch berücksichtigt, dass man eine Abschirmung der Kernladungszahl annimmt. Die Lösungen werden wasserstoffartig angenommen, d.h. man ignoriert die WW der Elektronen untereinander, es findet keine Deformation der Lösungen durch die Anwesenheit weitere Elektronen statt.
Dadurch erhält man einen Satz von Eigenzuständen (die die Chemiker Atomorbitale nennen).

Diese Lösungen passen zur Rotationsinvarianz eines isolierten Atoms.
Nun hat man jedoch eine andere Symmetrie zu betrachten, nämlich die eines speziellen Moleküls. Anstatt nun die Schrödingergleichung
|\psi\rangle = 0 )
für dieses extrem komplizierte Problem zu lösen, setzt man näherungsweise die Lösungen aus einzelnen Atomorbitalen zusammen:

Die Koeffizienten sind so gewählt, dass eine grundsätzliche Antisymmetrisierung der Zustände sowie eine an die spezielle Symmetrie des Moleküls angepasste Form berücksichtigt wird; außerdem geht natürlich die Anzahl der zu verteilenden Elektronen ein. Man erhält also einen neuen Satz an näherungsweise gültigen Eigenzuständen (die die Chemiker Molekülorbitale nennen).
Die Energie-Aufspaltung erklärt sich dadurch, dass unterschiedliche Zustände mit unterschiedlichen Koeffizienten beitragen. Um das im Detail zu verstehen, müssen wir die gewählte Näherungsmethode genauer untersuchen.
_________________
Niels Bohr brainwashed a whole generation of theorists into thinking that the job (interpreting quantum theory) was done 50 years ago. |
|
|
frustudent
Gast
|
| frustudent Verfasst am: 05. Aug 2014 14:52 Titel: |
|
|
Servus,
Machen wir das ganze am H-Molekül:
Wenn 2 H-Atome unendlich weit weg sind, so sind die Elektronen im s-Orbital. Bringst du sie nun näher zusammen, so überlappen sich die Wellenfunktionen der H-Atome. Die Wellenfunktionen der H-Atome kannst du nun unterschiedlich zusammen addieren, so dass sie das gleiche Vorzeichen haben (somit gibt es eine Aufenthaltswahrscheinlichkeit der Elektronen zwischen den Atomkernen) oder mit unterschiedlichen Vorzeichen (die Aufenthaltswahrscheinlichkeit der Elektronen verschwindet zwischen den Atomkernen). Somit hast du 2 mögliche neue Orbitale.
Hier ist das ganze auch nochmal grafisch dargestellt:
w3-o.hm.edu/fb06/professoren/maier/analytik/Blaetter/N032_Wechselwirkung_b_BAneu.pdf |
|
|
TomS
Moderator
Anmeldungsdatum: 20.03.2009
Beiträge: 21469
|
| TomS Verfasst am: 05. Aug 2014 15:37 Titel: |
|
|
| frustudent hat Folgendes geschrieben: | | Hier ist das ganze auch nochmal grafisch dargestellt |
schöne Zusammenfassung; was jedoch fehlt ist die Berechnung der MO-Energie nach Anwendung der (gut dargestellten) LCAO; kennst du sowas?
_________________
Niels Bohr brainwashed a whole generation of theorists into thinking that the job (interpreting quantum theory) was done 50 years ago. |
|
|
frustudent
Gast
|
| frustudent Verfasst am: 05. Aug 2014 17:57 Titel: |
|
|
Ja, ist allerdings nicht mehr ganz so anschaulich.
Ich mache es mal an einem 2-atomigen Beispiel (H2, HCl, ...):
Wie schon gesagt, die Gesamtwellenfunktion wird als Linearkombination aufgeschrieben:

Die Koeffizienten und auch die Energie kann dann mit dem Ritzschen Variationsverfahren berechnet werden:

Betrachten wir den Zähler, so ergibt sich:

Meistens werden noch ein paar Kurzschreibenweisen verwendet:

Das ist die Energie des freien Atoms.

Dieser Term gibt dann die stärke der Bindung an.
Wenn man nun den Nenner auch noch mit auswertet, so bekommt man für die Energie:

Dabei ist S das sogenannte Überlappintegral:
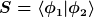
Wenn die Orbitale unendlich weit weg sind, ist das Überlappintegral 0, wenn man die näher zusammenführt, ändert sich es.
Die nötigen Parameter kann man ausrechnen, die Koeffizienten c1 und c2 müssen so bestimmt werden, dass die Energie minimal wird.
Vielleicht eine kurze Randbemerkung: Die LCAO Methode ist vielleicht anschaulich, liefert aber unglaublich schlechte numerische Lösungen. In der Quantenchemie werden daher heutzutage andere Methoden verwendet (Hartree-Fock, Atoms in Molecules: QTAIM, Dichtefunktionaltheorie,...). |
|
|
TomS
Moderator
Anmeldungsdatum: 20.03.2009
Beiträge: 21469
|
| TomS Verfasst am: 05. Aug 2014 23:20 Titel: |
|
|
Danke
_________________
Niels Bohr brainwashed a whole generation of theorists into thinking that the job (interpreting quantum theory) was done 50 years ago. |
|
|
TomS
Moderator
Anmeldungsdatum: 20.03.2009
Beiträge: 21469
|
| TomS Verfasst am: 06. Aug 2014 12:36 Titel: |
|
|
welche Methode wendet man an, wenn man nicht nur den Grundzustand finden möchte?
_________________
Niels Bohr brainwashed a whole generation of theorists into thinking that the job (interpreting quantum theory) was done 50 years ago. |
|
|
jh8979
Moderator

Anmeldungsdatum: 10.07.2012
Beiträge: 8762
|
|
|
TomS
Moderator
Anmeldungsdatum: 20.03.2009
Beiträge: 21469
|
| TomS Verfasst am: 06. Aug 2014 13:30 Titel: |
|
|
Hm, versthe ich nicht.
Die DFT benutze ich für stationäre Probleme, die TDDFT für zeitabhängige Probleme, das brauche ich doch gar nicht.
Sind die Kohn-Sham-Funktionen der DFT evtl. schon ausreichend? Umfassen die auch die angeregten, stabilen Zustände?
_________________
Niels Bohr brainwashed a whole generation of theorists into thinking that the job (interpreting quantum theory) was done 50 years ago. |
|
|
jh8979
Moderator
Anmeldungsdatum: 10.07.2012
Beiträge: 8762
|
| jh8979 Verfasst am: 06. Aug 2014 13:31 Titel: |
|
|
Ich muss gestehen ich hab nur kurz drüber geguckt und das hier gelesen:
"... to investigate the properties and dynamics of many-body systems in the presence of time-dependent potentials, such as electric or magnetic fields. The effect of such fields on molecules and solids can be studied with TDDFT to extract features like excitation energies, frequency-dependent response properties, and photoabsorption spectra." |
|
|
TomS
Moderator
Anmeldungsdatum: 20.03.2009
Beiträge: 21469
|
| TomS Verfasst am: 06. Aug 2014 17:27 Titel: |
|
|
OK, danke; man muss das wohl mal selbst durchgearbeitet haben ;-)
_________________
Niels Bohr brainwashed a whole generation of theorists into thinking that the job (interpreting quantum theory) was done 50 years ago. |
|
|
|


